Each year, hundreds of thousands of people have their genomes sequenced to diagnose an illness, discover their ancestry, or learn about their genetic risk for certain diseases. Yet, almost none of those genomes are read from scratch. A sequencing machine reads a patient’s DNA in fragments, and software then lays each fragment against an agreed-upon sequence called the human reference genome. This reference acts like a master map, guiding the software to anchor the small fragments where they most likely belong. Without it, reading a genome would be slower and far more expensive.
However, for most of the past twenty years, that map has largely been one person: an anonymous man who answered a newspaper advertisement in Buffalo, New York, in 1997, gave a blood sample, and was told in writing that he could expect no more than a tenth of the final sequence to come from him. About seventy percent of it did. As far as anyone involved knows, he has never learned that his DNA underlies most of human genetics, and the people who could find him have decided not to. The reference is not the human genome the phrase implies. It was assembled under deadline pressure, shaped by the engineering constraints of the time and by the politics of a project that spent its last years racing a private rival. And the choice of whose DNA became the default is not just an ethical matter. It follows the sequence into the clinic. The further a patient’s DNA sits from the reference, the worse the machine reads it.
- 1990Project launchesNIH and the Department of Energy begin a roughly $3 billion international consortium to read all 3.2 billion bases.
- 1992Watson resignsThe project’s first director quits over the NIH’s attempt to patent raw gene fragments.
- 1996Bermuda PrinciplesThe consortium agrees to post every base of public sequence online within 24 hours, before anyone can patent it.
- 1997RP11 recruitedA Buffalo News ad draws twenty donors; one anonymous man's library will supply about seventy percent of the reference.
- 1998The race turns to warCraig Venter founds Celera, promising the genome in three years; the public project pulls its finish line forward to 2003.
- 2000Working draftClinton and Blair announce a draft at the White House, "the human genome," singular.
- 2001Dual papersThe consortium publishes in Nature and Celera in Science, after a basement-pizza truce.
- 2003"Finished" sequenceDeclared complete on the 50th anniversary of the double helix, though the heterochromatin stayed unread.
- 2007Reference ConsortiumStewardship passes to the Genome Reference Consortium; Illumina acquires Solexa's short-read chemistry.
- 2009GRCh37 / hg19Becomes the coordinate system for nearly all of human genetics, with hundreds of gaps still in it.
- 2013GRCh38 / hg38Fixes thousands of errors and adds alternate contigs, which quietly break alignment in the immune loci.
- 2022T2T-CHM13The first gapless genome, from a mole cell line with no maternal DNA to phase, adds nearly 200 million bases.
- 2023Y chromosome & first pangenomeHG002 completes the Y; the Human Pangenome draft delivers 47 diploid genomes, 94 haplotypes.
- OngoingToward a pangenomeThe consortium aims for 350 individuals and 700 haplotypes, a reference meant to belong to everyone.
The public didn’t know any of that at the time, though. When the draft was later unveiled at the White House in June 2000, Bill Clinton in the East Room and Tony Blair by satellite from London, the phrase that carried was “the human genome,” singular, as though the project had read out a sequence belonging to everyone equally. A finished version followed in 2003 with similar fanfare. What the consortium had in fact produced was a composite: a mostly haploid path that flattened each person’s two inherited chromosome sets into one and drew the bulk of its sequence from that one donor in Buffalo.[1] How a sequence announced as everyone’s came to be mostly his is a story that begins a decade earlier, before there was any race to win.
The Genome War
The idea of sequencing the entire human genome came from the Department of Energy, which had spent the postwar decades studying how radiation damages DNA and wanted a complete reference against which to measure mutation.[2] The National Institutes of Health (NIH) soon took the lead, and in 1990 the two agencies launched a roughly $3 billion effort to be run as an international consortium. By its completion, the International Human Genome Sequencing Consortium spanned twenty centers across the United States, the United Kingdom, France, Japan, Germany, and China, and its 2003 finish was marked by a joint proclamation.[3] China joined last in 1999, founding what became BGI to sequence the tip of chromosome 3 as the only participant from a developing country.

Figure 1: Applied Biosystems 3700 capillary sequencer, the workhorse that produced the reference sequence at the turn of the century. These machines ran 96 capillaries in parallel, reading traces off fluorescently labelled Sanger products, and their throughput set the scale of the Human Genome Project.
[Courtesy of the National Human Genome Research Institute (NHGRI)]
The project’s first scientific director was James Watson, who quit in 1992 after a public fight over patents. A researcher in Craig Venter’s lab at NIH had been generating short gene fragments called expressed sequence tags, and the NIH, under director Bernadine Healy, had filed to patent them. Watson considered the patenting of raw genetic information indefensible. He resigned over the argument, and Francis Collins, a physician-geneticist, took over the public effort in 1993. Watson’s successor as NIH director, Harold Varmus, eventually abandoned the patent applications, but that act set the terms of everything that followed.
The public project answered that question largely through the conviction of one man. John Sulston ran the British end of the consortium at the Sanger Centre, funded by the Wellcome Trust, whose money made the United Kingdom the second great power of the effort and a counterweight to American commercial interests. Sulston had come to the human genome by way of a worm, the millimetre-long nematode C. elegans, whose every cell division he had traced in work that later won a share of the 2002 Nobel Prize. From the worm community he and his collaborator Robert Waterston had carried a practice of pooling data daily, and they pressed it onto the human project. Under the Bermuda Principles, agreed at a 1996 strategy meeting, every stretch of publicly funded sequence had to be posted to the internet within 24 hours of being read, before anyone could file a patent on it.

Figure 2: John Sulston insisted every base of the public genome reach the internet within 24 hours, before it could be patented.
[Courtesy of Wellcome]
For Sulston it was close to a moral law. He believed profit had no business shaping what counted as basic knowledge of the species. But the rule was also a legal manoeuvre. Through the 1990s the United States Patent and Trademark Office treated isolated DNA as a patentable composition of matter, so a human gene, once cut from the genome, could be claimed like an invention, and companies had begun filing in bulk on raw gene fragments whose function no one yet knew, betting that a claim staked early would pay off once a use was found.[4] The consortium could not rewrite patent law, but it could dissolve the novelty those claims depended on: a sequence already in public view is prior art, and prior art cannot be patented. Posting every night pushed each stretch into the public domain before anyone could fence it off, a manoeuvre that even predated Celera, which would not exist until 1998. In practice the 24-hour deadline was often technically infeasible, so it acted more as an aspirational baseline the consortium strained toward, even when reality fell short.
Bacterial Cloning and the Repeat Problem
For all the politics, no one had yet assembled human DNA into a single, continuous genome. The obstacle that would later leave the reference full of holes was repetition: long stretches of the human genome are made of the same short unit copied over and over, and a sequencer that reads only a small piece at a time cannot tell one copy from another.
There was more than one way to assemble it, and the consortium chose to keep the problem small using an approach called clone-by-clone sequencing. Rather than shred the whole genome at once, it broke the genome into pieces of known chromosomal position, each roughly 150,000 to 200,000 bases long, and cloned each one separately before reading it. Confining the assembly to one small chromosomal window meant a repeat could only be confused with a copy sitting immediately nearby.
One such vehicle that can be used for this task is the bacterial artificial chromosome, or BAC, introduced by Hiroaki Shizuya and Melvin Simon at Caltech in 1992. A BAC is built on the F-factor, the fertility plasmid of Escherichia coli, whose partitioning machinery holds the construct to one or two copies per cell. That low copy number is what keeps a large human insert stable, giving the repeat-laden DNA few chances to recombine with itself. BACs had replaced the earlier yeast artificial chromosomes, which held larger fragments but frequently produced chimeras, two unrelated pieces of human DNA fused into a single false clone.
Figure 3: An example of a bacterial artificial chromosome (BAC) vector map. The F-factor backbone keeps the construct at one to two copies per cell, minimizing recombination between repeat-laden human inserts.
[Courtesy of Addgene]
Growing DNA inside bacteria can seem like the harder path when, by the 1990s, the polymerase chain reaction could copy DNA in a test tube. Kary Mullis had worked out PCR in 1983 while driving up the California coast toward Mendocino, an idea that won him a Nobel Prize a decade later.[5] But PCR was unsuited to genome-scale cloning for several reasons. The first was accuracy: Taq polymerase, the standard PCR enzyme, lacks the exonuclease domain that lets a cell proofread and excise a wrongly added base, and it misincorporates at roughly to per base. A bacterium copying its own chromosome pairs that proofreading with a downstream mismatch-repair system, driving the error rate to about to per base, far more faithful than the alternative. The other, more fundamental problem, is that PCR needs short primers to mark where copying begins, so the sequence flanking the target must already be known, and the whole point of the project was to read sequence no one knew yet. Random hexamer primers might sound like a plausible idea, until you realise they will introduce chimeras and GC-bias. Gluing an unknown human fragment into a BAC solved both at once: the bacterium copied the insert faithfully and researchers could prime from the vector’s familiar sequence rather than from the mystery within. Inserts in the library built for the project averaged 178 kilobases.
However, even inside a BAC, the stability had limits. When an insert held long arrays of near-identical tandem repeats, the copies recombined through the host’s homologous-recombination machinery, looping out and deleting the DNA between them. The standard countermeasure was to grow libraries in recombination-deficient host strains, such as mutants.[6] Even then the most repetitive regions stayed unstable and the libraries quietly shed them, which is why the centromeres and other satellite-rich stretches never entered the early reference.
Once you have a library of clones, reading the genome become the next challenge. It was painstaking and hugely expensive. Sanger sequencing, the best method of the day, ran out after about 500-800 bases. Meanwhile, our genome spans roughly 3.2 billion base pairs. Every clone had to be shattered into small fragments, each read on its own and stitched back together by its overlaps, with each base read eight to ten times over so the errors would cancel. That redundancy meant tens of billions of bases of raw data, tens of millions of separate reads, off hundreds of capillary machines like the one in Figure 1 running day and night for years. It is most of where the project’s $3 billion and thirteen years went.
To kick off the project, the consortium needed DNA from human donors. Where the project had been getting it though, is the part of the story the archived documents would later expose.
The behind-the-scenes account that follows, including the internal emails, the ethics-board vote, and the interviews drawn on below, was first reported by Ashley Smart in an investigation for Undark, co-published by STAT, which obtained more than 100 documents from the National Human Genome Research Institute’s History of Genomics Archive. The narrative here follows that reporting.
By 1996, the project had a problem. It had been building its clone libraries from whatever human DNA was at hand, and the archived correspondence Undark reviewed shows the pool had come to include a cadaver and blood from scientists on the project itself, most of it collected outside any formal consent process or ethics review. What unsettled Collins most was that for at least one library, the scientists handling the DNA knew exactly whose it was, and the donor himself knew it was being used. He and Aristides Patrinos, of the Department of Energy, drew up a new plan. The libraries that would supply the published genome would be made under a double-blind protocol, so that the people handling a library could not learn whose blood it came from, and a volunteer could never confirm that their own had gone into it. The final sequence, in turn, would be a mosaic drawn from many people, so that no single individual would be exposed or able to claim ownership. This decision was made to prevent donors from becoming media spectacles and to protect them from potential exploitation. After all, at the time, no one had ever had their entire genome sequenced, and the risks were entirely unknown. Caltech and the Roswell Park Cancer Institute in Buffalo were commissioned to build new libraries following these rigorous new standards.

Figure 4: A 1996 memo and a 1998 letter exposing the Human Genome Project’s early library problems: DNA collected without consent or IRB approval, from non-anonymous donors, and even from a cadaver. A handwritten note on the letter questions whether the informed consent it describes was ever valid.
[Visual: Undark]
Pieter de Jong, who ran the Roswell Park cloning effort, recruited his donors through a lottery. He advertised in the Buffalo News on 23 March 1997, the same edition running a front-page feature on the project.

Figure 5: The advertisement placed by Pieter de Jong in the Buffalo News on 23 March 1997, seeking twenty volunteers to donate blood for the Human Genome Project.
[Courtesy of the National Human Genome Research Institute’s History of Genomics Archive]
One by one that spring, twenty respondents, evenly divided between men and women, sat with a genetic counselor, put their names to a consent form, and had blood drawn. The counselor tagged each sample with a number and left nothing on paper that tied a number back to a name. The signed forms were then locked away in sealed envelopes. From the twenty, de Jong chose one man and one woman at random. The first library came from the male donor and was built mostly by a postdoc, Kazutoyo Osoegawa, who was unusually good at pulling long, intact pieces of DNA from a sample. The library, RPCI-11, or RP11, turned out unusually well-behaved: of 169 clones checked, only 11 percent carried even minor rearrangements, and not one was chimeric. Its first clones reached the sequencing centers in August 1997. The consent form those twenty had signed told them, in writing, that no more than about 10 percent of the eventual sequence was expected to come from any one donor.
The Race
In 1998, the race turned into open war. Venter had left NIH in 1992 to start a nonprofit institute, then joined with the instrument-maker Perkin-Elmer and its president Michael Hunkapiller to found a company, Celera Genomics. Celera announced that it would sequence the human genome in three years for about $300 million, finishing years ahead of the public timetable. Collins read the move as an attempt to have the public effort shut down, and the consortium responded by accelerating their own timeline. It set a new goal of a working draft by 2000 and a finished sequence by 2003 and raised its funding sharply. What let Celera promise three years where the consortium needed thirteen came down to the method each side used to read three billion bases.
Both efforts used shotgun sequencing, shearing the DNA at random and reassembling it from overlaps. What separated them was scale. The consortium worked one mapped clone at a time, the method known as hierarchical shotgun or clone-by-clone, which meant first building a physical map: fingerprinting thousands of clones and ordering them into a tiling path along each chromosome before any were read. That mapping took years and was the slowest part of the work, but it kept each assembly confined to a single 178,000-base window, where a repeat could be confused only with a copy nearby. Celera ran whole-genome shotgun on everything at once, with no map, quicker to begin but leaving a computer to untangle every repeat in the genome simultaneously, which the consortium feared would stitch distant repeats together by mistake. Skipping the map is how a private company could promise to finish a decade’s work in three, and the threat of being overtaken, the result then metered behind a subscription, is what turned a methodical project into a sprint.
That sprint led to the question of whose DNA the project would use. The 10 percent promise dissolved over the next two years. The original plan had called for ten new libraries to be blended together. The real reason behind the decision to abandon that plan is something its own leaders no longer agree on, as Undark found when it put the question to them. Collins and Eric Green recall the reasons as mainly technical. Blending ten genomes is error-prone, because structural differences between people, such as large insertions and deletions, make two sequences hard to mesh cleanly. To top it off, the RP11 library was simply the best they had. Its cloned fragments were unusually large, which made the surrounding genome easier to map, it sat closer to sequencing than any of the others. Waterston, de Jong, and Patrinos remember it differently. The decisive factor, they told Undark, was time. The deadline Celera had forced on the project left the other new libraries unfinished, while RP11 already was. There was no second library to turn to, so the project drifted toward drawing most of the final sequence from this one well-characterized donor, and the 10 percent promise went with it.
The records of how that promise was set aside, drawn from the emails and memos Undark obtained, show a project choosing speed over the promise it had made. In a September 1998 email to his deputies, Collins wrote that the consent form wording had been nagging at him since a colleague first noticed it. The form’s word “expect” did not strictly cap how much could come from one donor, he reasoned, and he wanted to know how far that limit could be stretched. He also did something unusual for a funder of research, sitting in on a call with the chair of Roswell Park’s ethics board. Within days the board unanimously agreed that the ten male volunteers would not be asked again, and RP11 could be used as widely as the work required. A form that said it “expected” a limit, the panel reasoned, had not guaranteed one. Reopening contact with the donors risked exposing who RP11 was, and holding back a project that stood to help so many patients would be the greater harm. Not everyone went along. Maynard Olson, who ran one of the sequencing labs, objected in writing, dismayed that the project was talking itself into treating a signed consent form as something less than a promise. Patrinos was blunter when Undark reached him years afterward. The team had been in a panic to finish, he said, and the ethics were simply not what anyone was thinking hardest about.

Figure 6: The Roswell Park consent form told donors no more than 10 percent of the genome was expected to come from any one of them. RP11 supplied roughly seventy.
[Courtesy of Undark]
Some place the episode alongside the HeLa cell line, taken from Henrietta Lacks without her knowledge and grown in labs all over the world ever since. But Lacks never signed any consent form, RP11 technically did. He answered an advertisement, sat with a genetic counselor, signed a form, and was promised both anonymity and a ceiling on how much of him would end up in the sequence. The ethical issue was more about the choice to reason around it once the wording in the form became inconvenient. Perhaps that is why the two stories have travelled so differently. RP11 stayed essentially unknown until the 2024 Undark and STAT investigation, largely absent from public scrutiny and bioethics courses.
The Truce
The contest between the two efforts reached the White House. On 14 March 2000, Clinton and Blair issued a joint statement that raw genome data should be made “freely available to scientists everywhere”. Investors misread it as a threat to gene patents, even though the statement explicitly endorsed them, and the misreading triggered a sell-off. Celera’s shares fell 22 percent in a single day on the New York Stock Exchange, and the slide continued for weeks, dragging tens of billions of dollars out of the biotechnology sector. Clinton, who wanted the feud ended, asked Collins to find a way out, and Collins turned to Patrinos, who was on friendly terms with both sides. Patrinos invited Collins and Venter to his house and sat them in his basement over pizza, where, across a few Sunday meetings, the two agreed to a truce. They would announce together and publish separately. The dual papers appeared in early 2001, the consortium’s in Nature and Celera’s in Science.[7] Patrinos later recalled how quickly the ice melted once the cameras were gone.

Figure 7: Craig Venter (left) and Francis Collins (right) at a June 2000 White House ceremony celebrating the working draft of the human genome.
[Visual: NHGRI/Flickr]
As the race wound down, the whole-genome shotgun gamble had paid off better than the consortium predicted. It would become the standard way to read a genome for decades. Whether Celera had truly proven it, though, became its own public quarrel. Because the consortium posted everything within 24 hours, Celera had been free to pull the HGP’s accumulating sequence into its own assembly, and in a pair of PNAS papers Waterston, Lander, and Sulston argued it had leaned on that data, roughly 60 percent of the reads and all of the mapping data by their count, so heavily that the 2001 result proved little about the method on its own. Celera rejected the charge, saying it had shredded the public data, to which it was entitled, and rebuilt its assembly from the mate-pair links in its own reads.
The consortium’s own 2001 paper had disclosed, with far less attention, where its sequence came from. A table listed RP11 as the source of just over 74 percent of the sequence, the project’s own count at the time, with seven other libraries supplying small remainders of between 1.6 and 4.3 percent each. By a later accounting his true share came out nearer the seventy percent cited earlier. What the paper did not say was that six of those eight named libraries were the very ones whose ethically tangled origins had set off the 1996 crisis: the libraries whose donors were known to project scientists, those suspected of having been built from project members’ own DNA, and one drawn from the cadaver of a 19-year-old who had died by suicide, whose family had donated his body to science but never specifically consented to its use in the project. One of those tangled sources traced to roughly 10 percent of the genome that came from de Jong himself. Short of donors and in a hurry back in 1993, he and a visiting collaborator had drawn each other’s blood, and he admitted as much to Undark, reckoning the reference to be around three-quarters RP11 and a tenth him. A 2010 analysis of the reference placed RP11’s ancestry as a mix of African and European and judged that he most likely identifies as Black or African American. He has never been informed of any of it. The architects of the project worried at the time that he might one day learn of his role and have a claim. De Jong lives outside Seattle now, where he still does a small trade in clone libraries. Earlier this year, to clear space, he finally disposed of three of the five libraries he had made for the project.
Ironically, Celera’s draft leaned just as heavily on a single person. Venter eventually admitted he was even the majority donor himself. The other donors included a former Navy medic who described his war in Vietnam as “MAS*H without the jokes,” a near-failing high-school student turned beach bum turned genomics entrepreneur, and, as it happens, once a professor at Roswell Park in Buffalo, the same institute that produced RP11. Both reference genomes, public and private, are dependent on a single man.
The mosaic was supposed to prevent exactly this. The plan had been to blend the sequence from many donors so that no individual could be identified in the result or stake a claim to it, and Waterston, who helped design it that way, put the intent to Undark plainly: “It’s your genome. It’s my genome. It’s representative of everybody’s genome.” What was published was nothing of the sort.
The Reference as Infrastructure
Once the public draft had been built, finished, and published, it became the coordinate system against which every later human genome would be read, referenced in knowledge bases, and written into clinical pipelines that turn a patient’s reads into a diagnosis. The donor underneath it, chosen under a deadline in 1998, was now embedded in all of it.
The Reference Changes Hands
Celera won the science, their method proved to work. But they lost the business. Its plan had been to sell subscriptions to its genome database, but the public consortium was giving the same sequence away for free. The subscriptions never came in as hoped, and in January 2002 Applera’s management forced Venter out over the company’s direction. By 2005 Celera had closed the subscription service and handed its data to GenBank, the public archive it had spent years working around. The company remade itself as a maker of medical diagnostics, and in 2011 it was bought by Quest Diagnostics for about $671 million.
Celera’s sequence went to GenBank in 2005 and stayed there, sitting in the dust. The only part that saw continued use was Venter’s own genome, which his team reworked into a standalone diploid assembly in 2007. The public sequence was the one that survived, and in 2007 its stewardship passed to the Genome Reference Consortium, a partnership of the National Center for Biotechnology Information, the European Bioinformatics Institute, the Wellcome Sanger Institute, and Washington University. It was charged with correcting and extending the assembly it inherited, so the sequence carried forward and RP11 remained its largest single source. Re-sourcing it would have meant rebuilding from scratch the thing the field had already written itself onto, and the question of whose DNA it was, settled by default in 1998, was never reopened.
Physical Gaps and Broken Coordinates
The consortium’s first build, GRCh37, released in early 2009 (mirrored by the UCSC as hg19), became the coordinate system for nearly all of human genetics. The build captured the euchromatin well, but the heterochromatin did not survive. GRCh37 carried hundreds of gaps and omitted exactly the regions the BAC-and-Sanger pipeline could never resolve, including the centromeric satellite arrays, the telomeric caps, and the short arms of the five acrocentric chromosomes. Each centromere was instead replaced by a placeholder, a fixed gap of roughly three megabases of undefined sequence written into all 24 chromosomes.
The gaps survived the decade of collapsing costs because the short-read machines that took over the field read only about 150 bases at a time, far too short to cross an array in which the same 171-base-pair unit repeats thousands of times in a row. Every new genome was read by aligning its short fragments to GRCh37, which fixed the build in place as the coordinate system the whole field worked in.
GRCh38 arrived in December 2013 to correct thousands of single-base errors, close gaps with WGS data, and add modeled centromere sequence. Inserting new sequence renumbered the map. A variant at a given coordinate in GRCh37 now sat at a different integer in GRCh38, breaking backward compatibility for annotated databases. Chain files record how two assemblies line up, and tools such as UCSC’s liftOver and CrossMap use them to carry coordinates and annotations from one build to the next. A liftover is only as trustworthy as the correspondence between the builds, and it degrades or fails in exactly the regions GRCh38 rewrote, leaving many clinical laboratories stranded on hg19 for the better part of a decade. For all that it repaired, GRCh38 still could not read the genome’s most repetitive regions. its centromeres were modeled rather than sequenced, and its satellite arrays were left as placeholders.
Completing the Genome
Reading them at last meant a read long enough to cross an entire array of repeats and anchor in the unique sequence beyond, which neither Sanger nor the short-read machines could produce. Two sequencing technologies that matured in the 2000s would later allow us to finally have a complete genome.
Pacific Biosciences watches a single polymerase copy a strand inside a well small enough that the tracking light reaches only the enzyme at its bottom, recording each base as it is added. A single pass is error-prone, but circularizing the template and reading it over and over averages the mistakes out, and this circular-consensus mode, sold as HiFi, yields reads of about 13.5 kilobases at 99.8 percent accuracy.
Oxford Nanopore reads DNA with no polymerase at all. It draws a single strand through a protein pore held under a voltage, and because the four bases differ in size, each one disturbs the ionic current by a different amount as it passes, a signal the instrument decodes into sequence. The idea came to the biophysicist David Deamer on an Oregon roadside in 1989 and took more than a decade of others’ work to become the portable MinION sequencer, which reached users in 2014.[8] Its strength is raw length, with continuous reads beyond 100,000 bases demonstrated in 2018, far more than enough to cross a centromere.
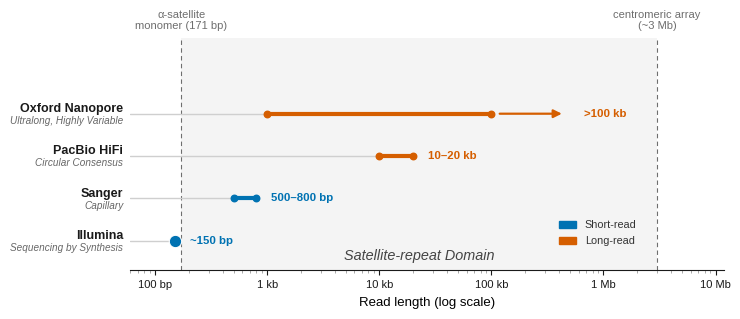
Figure 8: Sequencing read length against the scale of repeats, log axis. For two decades, reads were shorter than centromeric satellite arrays. Long reads extended by two to three orders of magnitude, enough to anchor across them. A complete ~3 Mb centromere (a fixed gap in GRCh37) is still spanned by assembling many ultralong reads.
Each read type did what the other could not. Nanopore’s ultralong reads crossed the satellite arrays that had broken every earlier method. Meanwhile PacBio HiFi, which had better accuracy at the time, refined the sequence inside them.
T2T-CHM13 and the Motherless Genome
The 2018 ultralong-nanopore work convinced two researchers that a complete genome was finally within reach. Karen Miga, a satellite-repeat researcher at UC Santa Cruz who had spent the better part of a decade trying to get real centromere sequence into the human genome, and Adam Phillippy, a bioinformatician at the National Human Genome Research Institute, founded the Telomere-to-Telomere consortium in 2019 with Evan Eichler at the University of Washington.
Long reads solved the length problem, but left a second one untouched: ploidy. A normal human cell is diploid, carrying two versions of each chromosome that differ at millions of positions, and an assembler reconstructing a repeat-rich region has to decide, at every heterozygous site, which variant belongs to the maternal copy and which to the paternal. That decision is called phasing. Wrong phasing can shred an otherwise clean assembly into a tangle of switched and merged haplotypes.
To sidestep the phasing problem, the consortium chose a source with almost nothing to phase. It settled on the CHM13 cell line, derived from a complete hydatidiform mole, a non-viable conception in which the maternal chromosome set is lost and a paternal set is duplicated, so the tissue carries two copies of a single haploid genome and essentially no maternal DNA. CHM13 has a karyotype but is, for the purposes of assembly, uniformly homozygous.
The consortium had rehearsed the approach, finishing the human X chromosome in 2020 and chromosome 8 in 2021. Working from both read types, the 2022 assembly reached 3.055 billion base pairs and added almost 200 million absent from GRCh38, predicting nearly 2,000 new genes, of which about 99 appear to be protein-coding. For the first time it resolved the centromeres, where companion work by Nicolas Altemose and colleagues mapped the 171-base alpha-satellite monomers stacked into the higher-order repeats that organize the kinetochore. Pericentromeric and centromeric satellite together come to roughly 6 percent of the genome, hidden since 2000. Phillippy called the achievement the last base camp before the summit.
The one thing the mole could not supply was a Y chromosome. Its karyotype left the assembly complete across every chromosome but that one, so the Y required a second donor. In 2023, Arang Rhie and colleagues assembled it from HG002, a cell line long used as a benchmark, adding more than 30 million base pairs absent from the GRCh38 Y and finishing the last human chromosome.[9] The two efforts together produced T2T-CHM13v2.0, the first end-to-end reference spanning the full chromosome set at last.
Towards the Pangenomic Era
T2T-CHM13 is indeed complete, but only in a narrow sense. It is a finished sequence of one genome that is more thorough than the previous builds. But, just like GRCh38, it remains a single linear haploid string carrying one set of alleles, and aligning genomes from diverse populations to any one linear sequence produces reference bias. A read carrying a large insertion the reference lacks has nowhere to map, so the aligner files it as unmapped, making structural variants invisible. This bias is especially prominent in individuals whose ancestry differs the most with the reference.
From one donor to a population
Recruited at the Roswell Park Cancer Institute in 1997, the donor known as RP11 supplied a cloning library so stable it came to dominate the assembly. Consent forms reportedly expected no donor to exceed about 10 percent.
To be fair, GRCh38 had already tried to fold some of that variation into the reference while staying linear. But the attempt failed, sort of. For loci too variable to capture as one linear path, the consortium added alternate contigs, extra scaffolds holding divergent versions of the same region. The clearest case is the human leukocyte antigen (HLA) region on chromosome 6, the most polymorphic locus in the genome, with tens of thousands of known alleles. These contigs sit alongside the primary assembly without being connected to it. A read from a patient’s HLA locus can then match the primary path and an alternate contig equally well, and an aligner that records its confidence as a mapping quality has no basis to choose between them, so BWA-MEM assigns the read the lowest possible score, zero. Variant callers discard such reads by design to suppress false positives. Aligning to GRCh38 without special handling therefore throws away real reads and misses the variants on them, a loss of sensitivity in precisely the immune loci the contigs were meant to capture. The fix, making the aligner alt-aware and restoring meaningful mapping qualities afterward, arrived only years after the build.
It is clear that we need a better way forward. Fortunately, the limitations of a single reference string were identified in bacteriology years earlier. In 2005, Hervé Tettelin and colleagues coined the word “pan-genome” for a Streptococcus species, having found that any single isolate held only about 80 percent of the species’ genes and that new genes kept turning up with each strain sequenced. The Human Pangenome Reference Consortium applies the idea to people, and it is run partly from the same building as T2T, by Benedict Paten and Karen Miga at UC Santa Cruz with the coordinating center under Ting Wang at Washington University. Instead of a linear string, a pangenome is a bidirectional sequence graph, built for the human draft with the minigraph-cactus pipeline. Sequence shared across people forms the nodes. Where individuals differ, the graph branches into parallel paths, one per allele or structural variant, and rejoins downstream. Any one person’s genome becomes a path traced through the graph. The topology dissolves the alt-contig penalty. Graph-aware mappers such as Giraffe evaluate each read against many population haplotypes at once.
The people in the first draft, published in May 2023, came from the 1000 Genomes Project. The project had collected lymphoblastoid cell lines, immortalized white blood cells that supply an essentially unlimited source of a person’s DNA, banked at the Coriell Institute and drawn from 26 populations across five continental ancestry groups.[10] Crucially, those samples had been consented for unrestricted public release, which let the pangenome be built and shared in the open. The project had also sequenced hundreds of mother-father-child trios which allowed phasing to be possible.
The HPRC also took on the diploid problem that T2T had sidestepped. The draft used trio binning. The parents of each donor are sequenced with cheap short reads, the software then extracts the k-mers that are unique to the maternal genome and those unique to the paternal one. These haplotype-specific markers, called hap-mers, are then used to sort the donor’s own long reads into a maternal pile and a paternal pile before assembly begins. The approach is reference-free, avoiding the errors that often creep in when an assembler collapses both haplotypes into a single consensus and links variants that never occurred together. Where parental samples are unavailable, long-range data from Hi-C, which captures which stretches of DNA sit close together in the folded chromosome, or from Strand-seq, which preserves the directional identity of each homolog, supplies the phasing data. From 47 such donors the consortium assembled 47 phased, diploid genomes, 94 haplotypes in all.
Its composition inverts RP11’s legacy almost exactly. About half of the individuals had African ancestry, most of the rest came from the Americas and Asia, and only one had European ancestry. Against GRCh38 the draft added 119 million base pairs of euchromatic sequence, about 90 million of it from structural variation. Run on ordinary short-read data, the graph cut small-variant errors by 34 percent and roughly doubled the structural variants detected per haplotype compared with GRCh38 workflows. The consortium intends to keep expanding, toward 350 individuals and 700 haplotypes.
Why the Old Reference Persists
The project’s $3 billion price tag and thirteen-year timeline have both since collapsed. A genome read today against the existing reference costs around $200 and about a day, and even building one from scratch, the de novo route the project itself had no choice but to take, has fallen nearly as far. Long-read sequencing covers a human genome for a few hundred dollars at modest coverage, and an assembler turns the reads into a draft in hours on a single computer. What once took a global consortium thirteen years is now a few days’ work for one lab.
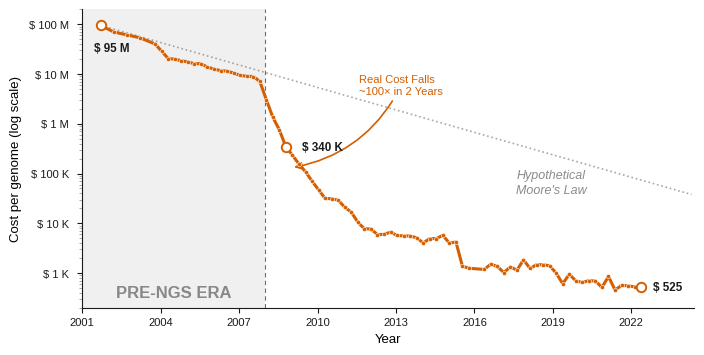
Figure 9: Cost per human genome, 2001-2022 (log scale). Sequencing tracked Moore’s law until next-generation sequencing arrived in 2008, then dropped ~100x in two years to roughly two orders of magnitude below it.
[Data: NHGRI Genome Sequencing Program, Wetterstrand KA, genome.gov/sequencingcostsdata]
So producing a better reference is no longer the obstacle. T2T-CHM13 is more complete than GRCh38, and the pangenome is more representative than either, yet the reference running in clinics and most pipelines is still GRCh38, released in 2013. It persists not because it is better but because the reference long ago became infrastructure, and the dependency runs the length of the pipeline, from the sequencer that reads a sample to the databases that turn it into a diagnosis.
At the sequencing end, the process that made genomes cheap also makes the reference indispensable. A clinical lab reads a patient’s genome in short Illumina fragments, the sequencing-by-synthesis method of David Klenerman and Shankar Balasubramanian commercialized through Solexa and bought by Illumina in 2007, which drove the cost of a genome down a hundred-thousand-fold in twenty years.[11] But those reads run only about 150 bases, too short to assemble across a repeat: the De Bruijn graph that short-read assemblers rely on folds every copy of a repeat onto the same nodes and cannot tell how many times to loop. So the reads are aligned to the reference rather than assembled on their own, and the reference tells each fragment where it belongs. And short reads are what nearly every lab produces, barring some labs that need the specific features of long-reads sequencing.
At the other end of the pipeline, the reference is part of the data itself. Every read and every variant is recorded by its position on it, so that coordinate is built into every file format. Aligned reads sit in BAM or CRAM files, and CRAM compresses against the reference so tightly that the data cannot be read back without the matching assembly. Variants sit in VCF files, each line a position and an allele. The databases that turn a variant into a diagnosis use the same coordinates, from gnomAD’s 807,162 individuals to ClinVar and dbSNP. Changing the reference shifts every coordinate, so the whole annotation layer has to be re-mapped and re-validated.
Validation has proved to be costly, which is why migration lags even between the two linear builds. Eight years after GRCh38 shipped, a 2021 survey found only 7 percent of clinical labs had fully adopted it and 54 percent had no plans to, citing cost and a lack of staff and validation resources. The American College of Medical Genetics has since had to publish formal guidance on how to make the switch. T2T-CHM13 cuts false-positive calls in 269 medically relevant genes by up to twelve-fold but renumbers the genome again. The pangenome goes further, abandoning the integer coordinate for a path through a graph that almost none of the installed clinical software can read, so its gains reach only the few labs already equipped to use it.
Which sequence got locked in was decided by how the war ended. Sulston’s 24-hour rule made the data impossible to patent, and Celera’s subscription business couldn’t survive against a free version of the same thing. That same open release pushed the public sequence into databases and pipelines worldwide before any alternative existed, and RP11 went with it, along with all the ethical issues we have discussed above. Ironically, the policy that kept the genome in the public domain is part of why a reference that represents almost no one, can no longer be cheaply replaced.
What holds the old reference in place is no longer the science. The more complete and more representative versions already exist. The only thing between them and the clinic is the labor of rebuilding everything written on top of the old one. So whose DNA stands in for the species is something the field now settles by default, the same answer reached under deadline in 1998: one anonymous man from Buffalo.
Footnotes
[1] The nuclear genome is not the only human reference that comes down to one person. The standard for mitochondrial DNA, the Cambridge Reference Sequence first published in 1981, was drawn mainly from a single individual of European descent, and the original even carried stray reads of bovine DNA and of HeLa cells, the line taken without consent from Henrietta Lacks. A corrected version in 1999 deliberately kept the old position numbering to avoid breaking every database built on it, the same lock-in that holds the nuclear reference in place. ↩
[2] The Department of Energy’s interest was not abstract. Its predecessor agencies had spent decades, through the Atomic Bomb Casualty Commission, trying to detect inherited mutations in the children of Hiroshima and Nagasaki survivors, a signal too faint for the methods of the day to resolve. Reading a report on that very problem in 1985, Charles DeLisi reasoned that a complete reference sequence was the way to measure such changes and pushed the idea into a DOE program the next year. ↩
[3] The timing was deliberate. The consortium declared the genome finished in April 2003 to fall on the 50th anniversary of Watson and Crick’s double-helix paper, which Nature had published in April 1953. Watson, the project’s first director, was a co-author of that paper. ↩
[4] The legal ground shifted only much later. Patent law bars claims on a “product of nature,” but the Patent Office had reasoned that a gene extracted, isolated, and purified in the lab was no longer natural. It was a novel composition of matter, no different in kind from a synthesized chemical or a new plastic. By the height of the genome race the government had already granted patents covering thousands of human genes, and Celera, too, would soon be filing provisional patent claims on thousands more. The Clinton administration had no authority to order the Patent Office to set that precedent aside; undoing it required an act of Congress or a ruling from the courts, and neither could move on the timescale of the race. The reversal came only in 2013, when the Supreme Court held in Association for Molecular Pathology v. Myriad Genetics that naturally occurring isolated human genes are not patent-eligible, though synthetic cDNA still is. The nightly release was the only lever the consortium could pull in real time. ↩
[5] Cetus paid Mullis a $10,000 bonus for the idea, while the rest of the PCR group reportedly received the company’s customary one dollar each. Cetus then sold the patents to Roche for $300 million, and Roche has collected billions in royalties since. The heat-stable enzyme that made the method practical had been scooped for free from a Yellowstone hot spring, and the park has never shared in the proceeds. ↩
[6] RecA catalyzes the strand-exchange step of homologous recombination. Disabling it in the host strain suppresses, but does not abolish, recombination between repeat copies. Replication slippage and residual pathways still delete the most unstable arrays, which is part of why centromeric and other satellite DNA never made it into the early reference. ↩
[7] The split ran deeper than the choice of journal. The consortium lodged its sequence in GenBank, the free public archive, as the Bermuda rules required. Celera instead kept its own on a company website that academics could read only under restrictions, with downloads capped at a fraction of a percent per person per week and redistribution forbidden. To publish the paper at all, Science waived its long-standing rule that sequence data be deposited in a public database, a concession that drew open protest. Celera handed the data to GenBank only in 2005, once it abandoned the subscription business. ↩
[8] The early R9 pore had a single sensing point, and because a run of identical bases barely changes the current, accuracy fell off for homopolymers of about five bases or longer. The R10 pore added a second constriction behind the first and resolves homopolymers up to roughly ten bases. The production pore is engineered from the E. coli CsgG channel, and a motor protein ratchets the strand through one base at a time while a microchip records the current. ↩
[9] HG002 makes a pointed contrast with RP11. The cell line comes from an Ashkenazi Jewish man enrolled in the Personal Genome Project, and it became one of the most heavily used benchmarks in genomics precisely because its donor consented broadly to open, public, and even commercial use. The last piece of the finished human reference came from someone who agreed to exactly what RP11 was never asked. ↩
[10] The cell lines are not perfect stand-ins. Lymphoblastoid lines are white blood cells made to grow indefinitely by infecting them with Epstein-Barr virus, and during that immortalization and the culturing that follows they pick up mutations and rearrangements of their own, changes that belong to the dish rather than the donor. Builders of reference material have to track them so that an artifact of the culture is not mistaken for human variation. ↩
[11] The fall was steep enough to outrun the chip industry. For years the cost of sequencing a genome roughly tracked Moore’s law, the doubling of computing power about every two years, but around 2007, as short-read machines arrived, it began dropping far faster, which is why the NHGRI plots its cost curve against a Moore’s-law line for comparison. ↩